






本文通过解读USP-NF <381>,为注射用药物包装中弹性体密封件的选择和合规性验证提供思路。
一、USP <381>迎来修订
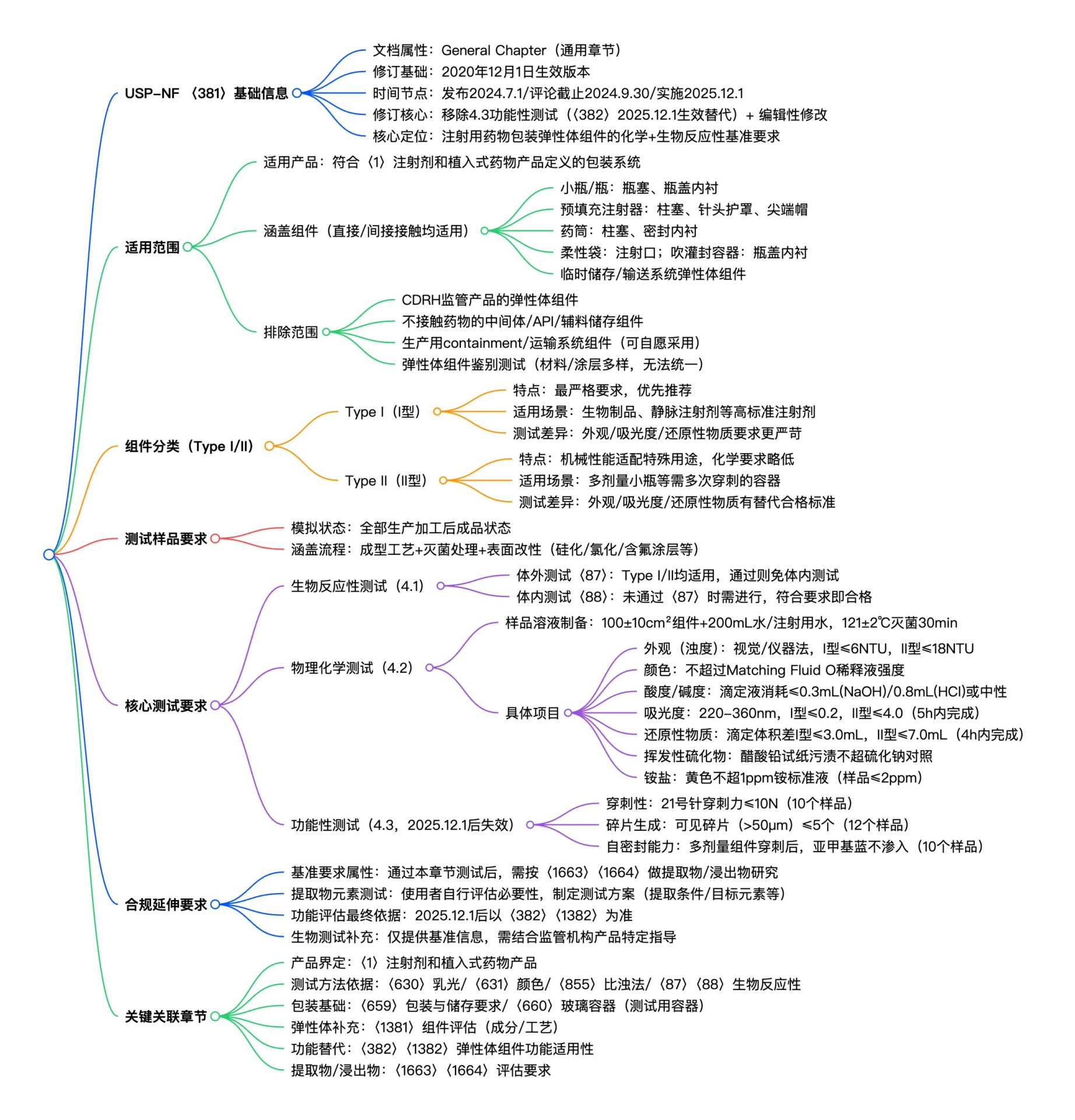
美国药典(USP)<381>章节,全称《注射用药物包装/输送系统中的弹性体组件》,是指导注射剂包装系统组件选型与合规验证的重要参考。该章节基于2020年12月1日生效版本进行了修订,于2024年7月1日发布,将于2025年12月1日正式实施。
此次修订的核心目的在于:
- 移除重复内容: 移除4.3“功能性测试”章节,避免与2025年12月1日生效的〈382〉章节内容重复。
- 规范格式: 进行编辑性修改,以适配当前USP格式规范。
修订后的<381>章节,将继续为注射用药物包装/输送系统的弹性体组件提供化学和生物反应性基准要求,是组件选型与合规性验证的重要指导。
二、适用范围与组件分类
1.适用范围
适用产品:符合《注射剂和植入式药物产品》〈1〉定义的产品包装系统。
涵盖组件类型:直接/间接接触药物的弹性体组件,包括:
- 小瓶/注射瓶用:瓶塞、瓶盖内衬;
- 预填充注射器用: plungers(柱塞)、针头护罩、尖端帽;
- 药筒用:柱塞、密封内衬;
- 柔性袋用:注射口;
- 吹灌封容器用:瓶盖内衬;
- 临时储存/输送系统中的弹性体组件(如多次穿刺型组件)。

排除范围:
- 医疗器械与辐射健康中心(CDRH)监管产品的弹性体组件;
- 不与药物直接/间接接触、仅用于中间体/API/辅料储存的弹性体组件;
- 生产过程中的弹性体组件(但可自愿采用本章节测试要求);
- 弹性体组件的鉴别测试(因材料与涂层种类繁多,无法统一制定方法)。
2.组件分类
USP <381>根据性能要求将弹性体组件分为两类:
| 类型 | 核心特点 | 适用场景 | 关键差异(测试要求) |
|---|---|---|---|
| Type I(I型) | 满足最严格要求,优先推荐使用 | 对化学纯度、稳定性要求高的注射剂(如生物制品、静脉注射剂) | 外观、吸光度、还原性物质测试要求更严苛 |
| Type II(II型) | 机械性能适配特殊用途,化学性能要求略低 | 需多次穿刺的容器(如多剂量小瓶) | 外观、吸光度、还原性物质测试有替代合格标准 |
选择哪种类型的组件,需要根据产品的特性和需求进行综合考虑。
三、测试样品要求:模拟真实状态
测试样品需模拟全部生产加工流程后的成品弹性体组件状态,包括:
- 成型工艺(如模塑条件)、灭菌处理;
- 表面改性处理(如硅化、氯化表面处理、含氟聚合物涂层、薄膜包覆等)。
四、核心测试要求:确保质量与安全
USP <381>对弹性体组件提出了严格的测试要求,主要包括生物反应性测试和物理化学测试。
1.生物反应性测试
两类组件均需通过体外或体内测试之一,以提供生物安全性基准数据。
2.物理化学测试
所有测试均需制备“样品溶液”和“空白溶液”(空白溶液不含弹性体组件,其余制备条件一致),样品溶液制备标准:
- 取总表面积100±10cm²的完整弹性体组件,放入硼硅酸盐玻璃瓶,加入200mL纯化水或注射用水;
- 121±2℃高压灭菌30min(20-30min内升温至设定温度),冷却后补充水分至初始质量,摇匀后立即收集使用。
| 测试项目 | 测试方法 | 合格标准(Type I / Type II) |
|---|---|---|
| 外观(浊度/乳光) | 视觉对比法(〈630〉)或仪器法(〈855〉),采用福尔马肼(Formazin)标准混悬液校准 | 浊度不超过参考混悬液B(6 NTU)/ 参考混悬液C(18 NTU) |
| 颜色 | 与标准比色液(Matching Fluid O稀释液)对比 | 颜色强度不超过标准比色液 |
| 酸度/碱度 | 以溴百里酚蓝为指示剂,用0.01N氢氧化钠或盐酸滴定 | 消耗滴定液不超过0.3mL(NaOH)/ 0.8mL(HCl),或呈中性 |
| 吸光度 | 0.45μm滤膜过滤后,在220-360nm波长下测定 | 吸光度≤0.2 / ≤4.0 |
| 还原性物质 | 样品溶液与高锰酸钾反应后,用硫代硫酸钠滴定 | 滴定体积差≤3.0mL / ≤7.0mL |
| 挥发性硫化物 | 20±2cm²组件与柠檬酸溶液混合,高压灭菌后通过醋酸铅试纸检测 | 试纸黑色污渍强度不超过对照溶液(硫化钠标准液) |
| 铵盐 | 采用碱性四碘汞酸钾试剂显色,与铵标准溶液对比 | 黄色深度不超过1ppm铵标准液(样品中铵含量≤2ppm) |
五、合规延伸要求:更全面的评估
通过USP <381>测试只是一个基准,还需要进行更全面的评估,以确保弹性体组件的实际适用性:
- 本章节测试仅为“基准要求”,弹性体组件通过后,需进一步按照〈1663〉(包装/输送系统提取物评估)和〈1664〉(药物浸出物评估)开展后续适用性研究;
- 提取物元素测试需由组件使用者根据实际使用场景(如提取条件、目标元素)自行评估必要性,并制定验证测试方案;
- 功能性评估最终需以〈382〉(注射用产品包装/输送系统弹性体组件功能适用性)和〈1382〉为准(2025年12月1日后正式替代本章节4.3);
- 生物反应性测试仅提供基准信息,具体产品的合规性需结合监管机构指导意见调整。
六、Type I组件符合性判断方法:三阶逻辑
结合USP-NF 〈381〉及补充摘要信息,判断弹性体组件是否符合Type I要求需遵循“基础属性确认→全项测试验证→合规延伸评估”的三阶逻辑,确保覆盖材料本质、性能指标与实际应用适配性。
1.第一步:确认Type I组件的基础属性与适用场景
在开展测试前,需先通过组件的材料组成、设计用途初步判断是否具备Type I的基础条件,避免无效测试:
-
材料组成要求:
Type I组件需以“经硫化(交联聚合)、加聚或缩聚形成的高分子有机弹性体”为基材,配方中可含天然/合成弹性体及无机/有机添加剂(如硫化控制剂、稳定剂、着色剂),但不得为纯硅酮弹性体(仅允许经硅酮处理,如涂覆二甲基硅油);
涂层需为“非阻隔性润滑涂层”(如化学/机械结合的润滑材料,不阻碍基材与药液的相互作用),若为聚四氟乙烯(PTFE)等阻隔性涂层,需额外验证基材本身是否符合Type I要求(不可通过涂层掩盖基材缺陷)。 -
适用场景匹配: Type I为“优先推荐使用的高规格组件”,仅适用于对化学纯度、稳定性要求极高的注射剂包装,包括:
- 水性制剂(如静脉注射剂、生物制品、疫苗);
- 长期储存的注射剂(符合〈659〉包装与储存要求);
- 无特殊机械性能需求的场景(如单次穿刺的单剂量小瓶),排除需多次穿刺、依赖特殊机械性能的多剂量容器(此类通常为Type II)。
2.第二步:全项通过USP 〈381〉规定的Type I专属测试要求
Type I的核心判定依据是通过〈381〉第4章“生物反应性测试”和“物理化学测试”,且需满足比Type II更严苛的合格标准,测试需严格遵循样品制备规范(模拟全生产流程后的成品状态),具体测试项目与判定标准见章节四。
3.第三步:合规延伸评估
通过〈381〉测试仅为“基准要求”,需结合以下延伸评估确认Type I组件的实际适用性:
提取物/浸出物评估:
- 按照《药用包装/输送系统提取物评估》〈1663〉开展受控提取研究,建立提取物谱(如挥发性有机物、半挥发性有机物、元素杂质)
- 按照〈1664〉在成品药液中检测确认浸出物,确保无超出安全阈值的迁移物(如元素杂质需符合ICH Q3D指南)。
材料一致性验证: 需确认组件的“身份属性”,包括:
- 尺寸、硬度、密度与供应商提供的Type I规格一致;
- 表面处理(如硅化)符合Type I要求,硅酮残留量需在规定范围内(≤10μg/cm²),避免影响药液澄清度或与API发生相互作用。
与药品的相容性验证:,包括:
- 长期和加速稳定性试验,监测组件的浸出物,是否对药液pH、澄清度、API含量产生不利影响;
- 吸附试验,确认组件不吸附药液中的API或辅料。
七、关键判定逻辑与常见误区
判定逻辑:Type I符合性需满足“基础属性匹配+全项测试通过+延伸评估合格”,缺一不可;若任一测试项目(如吸光度、还原性物质)未达到Type I标准,但符合Type II标准,则组件仅能归类为Type II,不可强行判定为Type I。
常见误区:
- 认为“通过生物反应性测试即可判定Type I”:需注意物理化学测试是Type I与Type II的核心区别,尤其吸光度、还原性物质的严苛标准不可省略;
- 依赖涂层掩盖基材缺陷:若组件为阻隔性涂层(如PTFE),需单独测试基材本身是否符合Type I要求,涂层不可用于“将不合格基材改造为合格Type I组件”;
- 忽略提取物/浸出物评估:〈381〉测试仅为基础,实际应用中需结合药液特性确认无迁移风险,避免因提取物导致药品稳定性下降或安全性风险。
参考文献
USP <381> Elastomeric Components in Injectable Pharmaceutical Product Packaging/Delivery Systems

