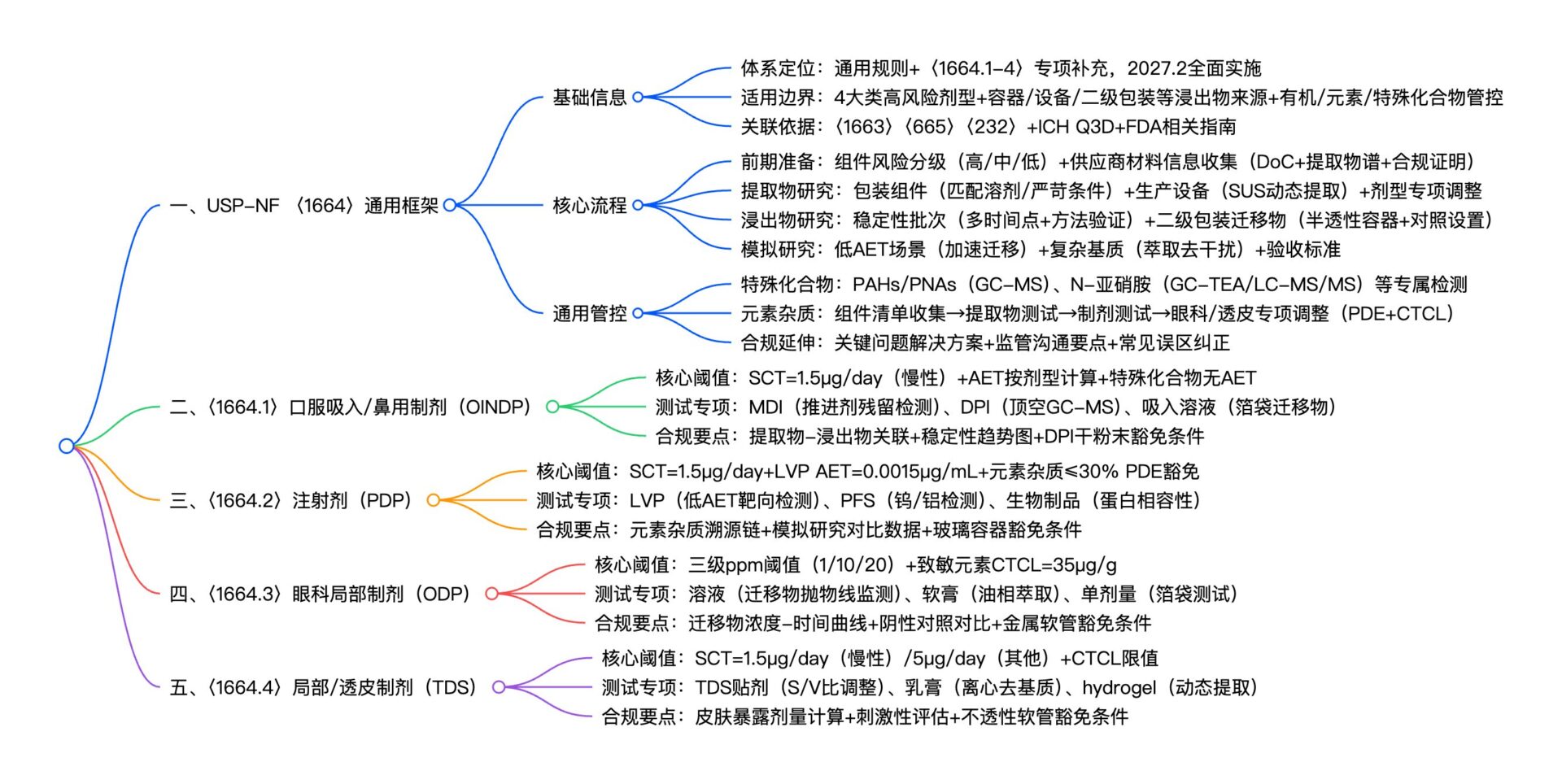
USP-NF 〈1664〉系列作为高风险药物浸出物管控的核心法规体系,以通用框架搭配四大专项章节(〈1664.1-4〉),聚焦口服吸入 / 鼻用、注射剂、眼科局部、局部 / 透皮四大高风险剂型,明确了浸出物从来源识别、测试流程到合规申报的全流程要求。
一、基础信息
| 维度 | 关键内容 |
|---|---|
| 文档体系定位 | 以〈1664〉为通用框架,〈1664.1-4〉为高风险剂型专项补充,构成“通用规则+剂型适配”的全链条浸出物管控体系,2027年2月全面实施(专项章节统一生效) |
| 核心适用边界 | – 覆盖剂型:口服吸入/鼻用(OINDP)、注射剂(PDP)、眼科局部(ODP)、局部/透皮(TDS等)4大类高风险剂型; – 浸出物来源:容器密封系统(CCS)、生产设备(含一次性系统SUS)、二级/三级包装(半透性容器场景); – 管控对象:有机浸出物、元素杂质(ICH Q3D规定)、特殊案例化合物(PAHs、N-亚硝胺等) |
| 核心关联依据 | 〈1663〉 、〈665〉 、〈232〉 、ICH Q3D(R2)、相关指南(如OINDP、相容性研究技术指导原则、生物制品包装指南) |
参考
USP <1664.1> 解读:OINDP 浸出物评估与合规策略
眼科用药安全:USP <1664.3> 浸出物评估与管控策略
皮肤给药制剂安全管控:USP <1664.4>浸出物管控全解析
二、核心内容
1. 提取物→浸出物→模拟研究
(1)前期风险分级
| 步骤 | 具体内容 | 参考 |
|---|---|---|
| 1. 组件风险分级 | 按“接触方式+剂型风险”分级: – 高风险:直接接触+高风险剂型(如MDI阀门、注射剂胶塞); – 中风险:直接接触+低浸出潜力剂型(如DPI泡罩); – 低风险:间接接触(如外层不透性标签) |
参考〈1664〉Table 1风险矩阵 |
| 2. 材料信息收集 | 向供应商索取3类文件: – 材料成分声明:明确聚合物、添加剂、金属杂质; – 提取物谱:历史批次的有机/无机提取物数据; – 合规证明:食品接触合规性(如21 CFR 174-186) |
建立《供应商材料信息档案》,作为测试方案设计依据 |
(2)提取物研究
| 测试对象 | 具体操作 | 剂型专项调整 |
|---|---|---|
| 单个包装组件 | – 提取溶剂:匹配制剂基质; – 提取条件:较存储条件严苛; – 提取用量:控制提取比例,确保达到AET浓度 |
– OINDP(MDI):增加有机推进剂模拟溶剂; – TDS:用模拟汗液(pH4.5-5.5)提取; – 注射剂:可采用安慰剂替代提取,需论证 |
| 生产设备(SUS) | – 提取对象:连接器、管路、过滤器; – 提取条件:模拟生产工艺(例如40℃、动态提取4h); – 检测重点:有机浸出物+金属杂质(如钨、铝、钴) |
– 生物制品生产设备:额外检测蛋白相互作用相关浸出物(如醛类、过氧化物) |
(3)浸出物研究
| 测试对象 | 具体操作 | 剂型专项调整 |
|---|---|---|
| 稳定性批次(≥3批) | 测试时间点: -常规储存:0、3、6、9、12、24个月; – 加速条件:40℃/75%RH(3、6个月); 方法要求:灵敏(LOQ≤AET)、专属(无基质干扰)、全验证 |
– ODP:需包含室温12个月迁移物测试(加速易假阴性); – 注射剂(LVP):增加中期时间点(如18个月); – TDS:同步测试贴敷期内浸出物迁移量 |
| 二级包装迁移物 | – 测试场景:半透性容器(如LDPE瓶、ODP滴管瓶); – 对照设置:无标签空白样品+完整包装样品平行测试 |
– OINDP(吸入溶液):包含箔袋内层迁移物测试; – 注射剂:标签/油墨迁移物 |
(4)模拟研究(低AET/复杂基质)
| 适用场景 | 具体操作 | 参考标准 |
|---|---|---|
| 低AET(如LVP AET=0.0015μg/mL) | – 模拟溶剂:匹配制剂“浸出能力”(如含表面活性剂的缓冲液); – 容器组合:完整包装(含二级包装)+ 模拟溶剂; – 测试条件:密封60℃储存4周(加速迁移) |
1. 识别≥AET的“可能浸出物”; 2. 毒理学评估显示无安全风险; 3. 作为靶向检测依据,替代部分制剂测试 |
| 复杂基质(如乳膏、生物制品) | – 预处理方法:液-液萃取(LLE)/固相萃取(SPE)去除基质干扰; – 空白对照:制剂安慰剂+提取溶剂空白 |
基质加标回收率80%-120%,RSD≤10% |
2. 分剂型专项实操要点
(1)关键阈值与测试方法适配
| 剂型 | 核心阈值 | 专属测试方法 |
|---|---|---|
| OINDP(〈1664.1〉) | – SCT=1.5μg/day(慢性),AET按剂型计算(如MDI=25μg/罐); – 特殊案例化合物:无AET,按检测方法LOQ管控 |
– MDI:冷过滤去除颗粒+ propellant venting残留检测; – DPI:挥发性浸出物用顶空GC-MS检测 |
| 注射剂(〈1664.2〉) | – SCT=1.5μg/day,LVP AET低至0.0015μg/mL; – 元素杂质:提取物≤30% PDE可豁免制剂测试 |
– 生物制品:额外检测浸出物诱导的蛋白聚集/氧化; – PFS:重点检测钨、铝等金属杂质(≤0.1μg/mL) |
| 眼科局部(〈1664.3〉) | – 三级浓度阈值:报告1ppm、识别10ppm、鉴定20ppm; – 元素杂质:加控致敏元素(Ni、Co,CTCL=35μg/g) |
– 半透性容器:室温12个月迁移物抛物线监测; – ointment:油相基质用正己烷萃取后检测 |
| 局部/透皮(〈1664.4〉) | – SCT=1.5μg/day(慢性)、5μg/day(其他); – TDS:模拟汗液提取(42±2℃,匹配佩戴时长) |
– TDS:表面体积比(S/V)按厚度调整(<0.5mm用6cm²/mL); – 乳膏:离心分离后取上清液检测 |
(2)特殊案例化合物管控
| 化合物类别 | 检测方法 | 备注 |
|---|---|---|
| PAHs/PNAs | GC-MS(选择离子监测模式) | 单个化合物≤LOQ |
| N-亚硝胺 | GC-TEA或LC-MS/MS | 符合〈1469〉规定 |
| 2-MBT | LC-MS | 橡胶组件专属 |
| PFOS/PFOA | LC-MS/MS | 覆膜胶塞场景 |
3. 元素杂质实操流程
- 收集组件材料清单:明确是否含 intentional 添加元素(如钴、镍);
- 提取物测试:按药典<232>方法测试组件可提取元素,结果≤30% PDE则豁免制剂测试;
- 制剂测试:高风险剂型(注射剂、生物制品)需在货架期终点测试元素杂质;
- 特殊调整:眼科/透皮剂型需同时满足PDE和CTCL(35μg/g),取较严值。
三、关键问题
问题1:ODP的二级包装迁移物呈抛物线变化,如何确定最大风险点?
解决方案:
- 增加中期密集时间点:0、3、6、9、12、18个月(首年每3个月测试);
- 环境控制:稳定性试验避免迁移物扩散;
- 判定标准:以“实测最大浓度”作为毒理学评估依据,而非终点浓度,需在注册资料中说明迁移规律。
问题2:注射剂LVP的AET=0.0015μg/mL,现有检测方法LOQ=0.01μg/mL,如何合规?
解决方案:
- 优化提取方案:增加组件用量(如20个LVP袋/250mL溶剂),提高提取物浓度至LOQ以上;
- 模拟研究补充:用匹配制剂浸出能力的溶剂开展模拟研究,识别可能浸出物并进行毒理学评估;
- 监管沟通:附方法验证数据+模拟研究数据,申请化合物专属阈值。
四、合规延伸
1. 监管沟通关键点
| 沟通场景 | 必备资料 | 沟通要点 |
|---|---|---|
| 低AET豁免 | 方法验证数据、模拟研究报告、毒理学评估报告 | 强调“提取物数据+毒理学”可覆盖风险 |
| 剂型特殊调整 | 剂型浸出物迁移规律数据、基质干扰验证数据 | 说明调整依据,符合法规精神 |
| 组件变更 | 新旧组件提取物对比数据、稳定性批次浸出物数据 | 证明变更后浸出物谱无显著差异,无新增风险 |
2. 常见合规误区
| 误区 | 后果 | 纠正动作 |
|---|---|---|
| 所有剂型统一用AET=1.5μg/day计算 | 阈值与剂型不匹配(如ODP无AET) | 按专项章节要求,OINDP/PDP用AET,ODP用ppm阈值 |
| 忽略生产设备浸出物(PERLs) | 生物制品出现蛋白聚集等质量问题 | 按〈665〉评估SUS |
| 加速条件替代长期条件迁移物测试 | ODP/半透性容器场景出现假阴性 | 迁移物测试必须包含长期数据 |
| 元素杂质仅测试制剂,未测试组件 | 无法追溯杂质来源 | 先测组件提取物,再按需测试制剂,建立溯源链 |