






随着一次性使用系统在生物制药行业的广泛应用,各国监管部门要求对其潜在浸出物进行评估。BioPhorum发布了浸出风险评估指南,概述了设计浸出研究时应考虑的关键参数。本文通过案例阐述了如何采用最佳实践根据工艺参数设计浸出试验,确保使用一次性系统后药品质量和患者安全。
在过去20多年中,一次性使用系统(Single-Used System,SUS)在生物制药制造中的使用显着增加,各国监管机构均出具法规和指南,要求制药企业评估和管理一次性系统对药品质量的风险。
浸出研究生成的数据可用于浸出风险评估,BioPhorum Operations Group(BPOG)制定的标准化提取测试方案有助于获取完整和可靠的数据。通过使用标准化协议生成完整的数据包,为制药企业在执行浸出风险评估时提供了重要信息。具体提取测试方案可参考相关文章“生产组件EL系列提取数据评估“。 此外,可根据需要对工艺中使用的一次性使用组件进行浸出研究,以完善对浸出风险的评估。
 下文从风险评估角度出发,介绍了BioPhorum关于浸出物风险评估的指南,以实际案例阐述浸出研究的设计。
下文从风险评估角度出发,介绍了BioPhorum关于浸出物风险评估的指南,以实际案例阐述浸出研究的设计。
1|浸出物风险评估指南
BioPhorum “BPOG”于2017年建立了一个基于科学的行业通用实践,以从浸出物的角度评估一次性使用系统对药品质量的风险,通过种风险评估的关键概念,为行业和监管机构提供了一个通用的方法。
BPOG《用于生物制药制造的聚合物单次使用系统浸出物风险评估最佳实践指南》(Best Practices Guide for Evaluating Leachables Risk from Polymeric Single-Use Systems Used in Biopharmaceutical Manufacturing),代表了来自17家大型生物制药公司的专家和小组的共识。该指南的目的是在采用单次使用系统的同时确保产品质量和患者安全。
指南分四部分,重点关注风险评估、浸出研究设计、分析方法以及从经验中汲取的教训。
2|风险评估模型
最佳实践指南中提供的风险评估模型与ICH Q9有关质量风险管理的指导意见保持一致,要求基于科学原则进行评估,最终与患者安全相关联。
通过风险评估模型,系统地将生产过程中的组件分为高、中、低风险类别。 根据风险类别确定所需的测试,决定特定组件质量控制过程所需的严格程度。在某些情况下,可提取物数据足以支持给定的SUS应用,不需要进一步的浸出测试;对于某些SUS则需要浸出测试以支持其应用。
有关风险评估的具体阐述详见文章“生产组件**E&L**系列 风险评估”。
3|浸出研究设计
根据风险评估结果,可能需要浸出研究以支持一次性使用系统(SUS)或组件组件的使用。在这些情况下,最佳实践指南概述了设计浸出研究时应考虑的关键参数,以确保稳健高效的研究,以确保稳健高效的研究。
设计浸出研究时应考虑的关键参数如表1所示(包括但不限于) 。
表1浸出研究设计关键参数
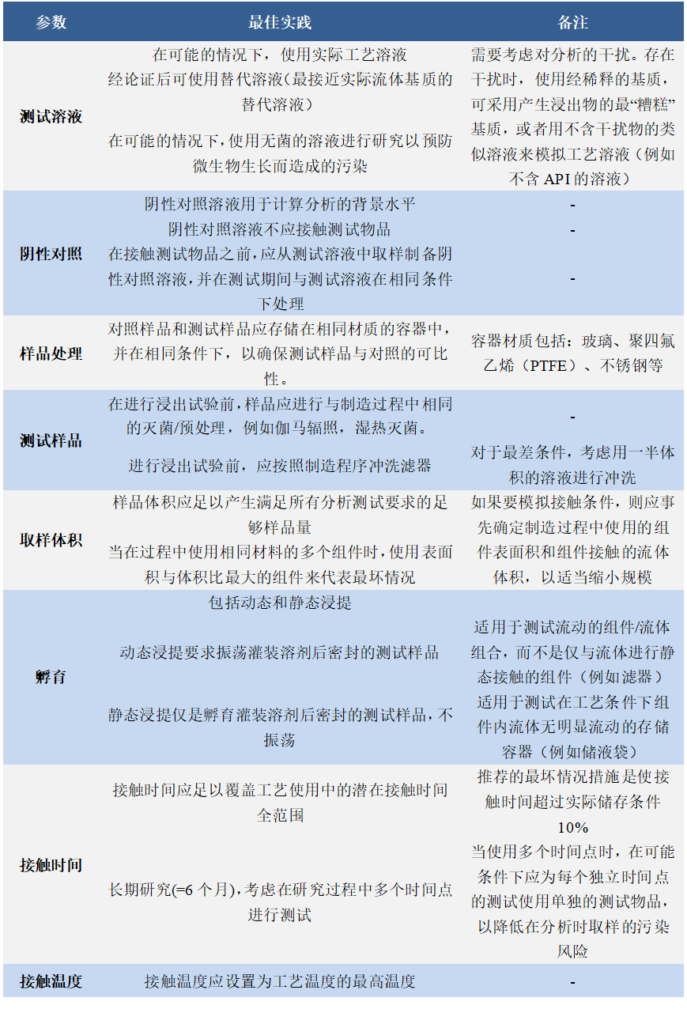 动态提取更适用于测试流通的组件/流体组合,而不是仅与流体进行静态接触的组件。
动态提取更适用于测试流通的组件/流体组合,而不是仅与流体进行静态接触的组件。
静态提取是在研究期间用已知体积的流体填充组件,密闭后对该测试物品进行孵化。静态条件适用于测试在工艺条件下组件内流体无明显运动的存储容器。提取参数应模拟实际工艺参数。
4|分析方法
需要使用可靠、稳健的分析方法来检测浸出物。最佳实践指南的分析方法部分概述了在开发浸出研究时应考虑的关键分析方法。
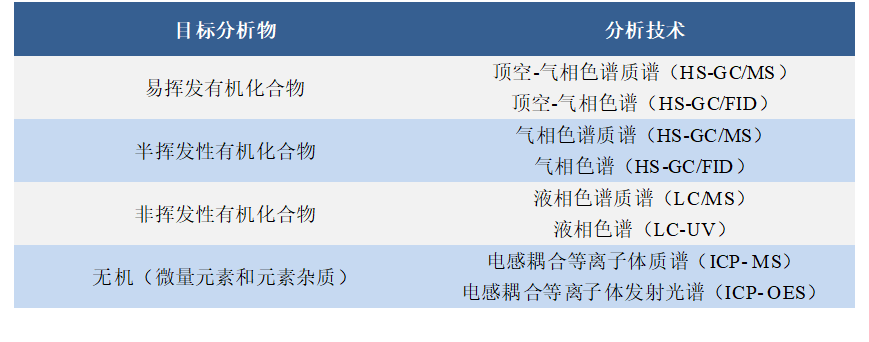
用于浸出研究的主要分析方法显示在表2中。
表2 浸出研究分析方法
5|案例研究
案例1: 生产工艺用配制袋
生物工艺袋用于生产过程中的配制和暂时存储,为多层复合膜,内层接触液体的材质为超低密度聚乙烯(ULDPE),体积从100L~ 1000L不等。配制的药品是pH接近中性的原液(含蛋白、表面活性剂、缓冲液等),操作温度16°C~ 30°C,与药品最长的接触时间为14天。根据最小灌装体积计算,最差情况下的袋子内表面积与体积比为0.12m2/L。
通过风险评估模型,确定了溶液与ULDPE生物工艺袋的接触为高风险。审查了ULDPE生物工艺袋的现有数据,没有足够的适用的可提取物/浸出物数据来支持该袋子的应用。因此,需要进行研究以验证生物工艺袋适用于批量药品配制和存储。
浸出研究的目的用于支持在ULDPE生物工艺袋中配制和存储药品。选择了两种浸提介质,分别为原液和和安慰剂。安慰剂与原液性质相似但不含蛋白质,避免蛋白存在带来的潜在分析干扰。
选择缩小规模的测试生物工艺袋,例如250ml袋,采用6cm2/L的比表面积比例进行试验。在使用之前,对生物工艺袋进行了伽马辐射,以保证与实际工艺一致的条件。
用测试溶液充满经辐射的测试生物工艺袋,并在40°C下保持14天、30天,以确保研究支持最坏情况的制造条件。同时取不接触工艺袋的溶液同时放置作为对照样品。
采用非特定化合物扫描方法,特定化合物物测试方法进行浸出物分析。
- 顶空-气相色谱质谱全扫描分析(HS-GC/MS):易挥发有机化合物
- 气相色谱-质谱全扫描分析(GC/MS):半挥发性有机化合物
- 液相色谱-质谱全扫描分析(LC/MS):非挥发性有机化合物
- 电感耦合等离子体质谱扫描(ICP- MS):微量元素和重金属(即无机元素杂质)
- 气相色谱-质谱 SIM分析(GC/MS):增塑剂、降解单挑等特定化合物
- 液相色谱-质谱 MRM分析(LC/MS/MS):抗氧剂降解物等特定化合物
在生物工艺容器中40°C下储存14天、30天后,扣除相应对照后,测试样品中检测到3种有机化合物(抗氧剂168降解物、支链烷烃)和4种元素(Si、Ca、Zn、Mg)。
对这3种有机化合物和4种元素的浸出量进行评估以确定其毒理影响。结合化合物的化学结构、药品使用途径和日摄入量,毒理学评估的结论是,根据所提供的浸出物数据,对患者安全性没有影响。
结论:ULDPE生物工艺袋适用于该制药生产工艺中批量药品配制和存储。
案例2: 上游工艺中使用的滤器
生产工艺中使用0.2微米聚醚砜(PES)过滤器过滤原料药。药液是pH接近中性的蛋白溶液。过滤操作在环境温度(<25°C)下进行,在12小时内完成。
对过滤器/工艺流体相互作用进行了风险评估,确定该过滤过程为中风险,现有数据不足以支持其应用。因此,需要进行研究以验证过滤器的适用性。
浸出研究所选测试溶液是在正常生产条件下过滤前的原料药,使用合格的过滤器进行试验,在使用之前参照工艺过程对过滤器进行冲洗和伽马照射。过滤器采用实际使用的型号,不需要缩小模型。提取比例为1:1(有效过滤面积:溶液体积),测试条件为30°C下24小时。同时取过滤之前不接触滤器的原料药作为阴性对照。
这里的案例是在实验室环境中使用受控条件制备测试样品,也可选择从生产线采集测试样品的方式。
同时分析对照样品和测试样品,经GC/MS、HS-GC/MS和LC/MS分析,均未检出超过AET阈值的有机化合物。ICP-MS结果显示测试样品中未检出微量或重金属。
结论:0.2微米PES过滤器被适用于生产工艺中原料药的过滤。
|总结|
根据组件的风险评级,当确定组件存在高风险时,可能需要进行浸出物测试,此前获得的提取物数据可作为预测浸出化合物的参考信息。对于最终灌装和包装工艺,通常具有较高的风险等级和更高的浸出物研究要求。
采用最佳实践可指导制药生产商进行风险评估,并合理设计浸出物研究,以确保在生物制药工艺中使用单用系统后确保产品质量和患者安全。
参考
-
Standardized Extractable Testing Protocol for Single-Use Systems in Biomanufacturing, Pharmaceutical Engineeringm 2014; 34(6), 1-11.
-
BPOG E/L Working Group: Best Practices Guide for Evaluating Leachables Risk from Polymeric Single-Use Systems Used in Biopharmaceutical Manufacturing. www.biophorum.com, 2017 3. BPOG 3. 3. 3.Extractables and Leachables Team, “Survey of Industry Leachables Best Practices Completed,” PDA Letter (March 2016).


