随着一次性使用系统(Single Use System,SUS)被集合到制药工艺中,可提取物和浸出物(Extractables& Leacheables)越来越接近最终的药物产品,成为制药行业需要考虑的关键问题。
生物制药过程的复杂性、组件系统材料和来源的多样性使得生产商面、制药行业带来了许多问题,包括可提取与浸出物相关法规要求;一次性生产组件的选择;使用期望值以及供应商能力等等。
有关SUS法规:
- 国内GMP: 规定“制造设备的材料不得对药品性质、纯度、质量产生影响,其所用材料需具有安全性、辨别性及使用强度。
- FDA 21 CFR 211.65 规定:确保与部件、过程中材料或药物接触的表面不会发生反应、添加或吸收,从而导致安全、性能、强度、质量或纯度超出官方或其他规定要求。
具体的实施指南还没有官方文档。
监管者和制药行业一直致力于制定标准化的提取和浸出物研究方案,希望能够涵盖整个过程中遇到的各种条件,并建议使用风险管理工具和基于质量源于设计 (QBD) 的理念来合理使用SUS系统。
参考国内外一次性系统生产组件相容研究的现状,伯朗氏实验室基于目前已完成的数百个品种的研究基础,从原则、概念、试验目的、合作等方面,分享一次性使用系统的 E& L研究的设计和实施经验。
关于更多的一次性使用系统的风险评估方法、提取试验设计、结果评估、浸出研究和案例,可以我们的公众号。
基于QbD原则
一次性使用的系统生产组件可沥滤物、浸出物的研究应基于QbD原则,尽可能全面地了解生物制造工艺。根据系统组件供应商提供的文件,首先评估组件对工艺流程的重要性和影响程度。评估的要素包括组件材料以下方面,作为开展相容性研究的信息收集基础
- 物理化学属性
- 法规符合性
- 预期用途和稳定性
- 药品的配方组成
- 预期临床使用方式和工艺条件
为了平衡企业的商业风险和患者安全,必须在 SUS生产和药物开发的不同阶段建立风险评估。考虑商业风险的同时不能以病人的安全为代价,主要考虑的因素必须是产品的质量和安全,并通过适当的风险评估和缓解策略,例如质量风险管理。 SUS生产商在满足终端用户(用户需求规范)方面投入了大量精力,每单位药物剂型的提取量(包括剂量学)是最终的监管预期,为了满足监管者的要求,制药企业必须遵守风险评估,以考虑患者的使用考虑。
SUS 可提取物和浸出物(E& L)定义
可提取物和浸出物反映出两种完全不同的化学成分,虽然两者都是由生产组件成分迁移而来。
可提取物:根据不同的实验室条件(如温度pH,极性和时间),由SUS向模型溶剂溶液中迁移的组件材料化学成份,相对于生物制药过程,该环境条件属于严苛条件。
浸出物:在常规制药工艺条件下从 SUS中迁移至工艺溶液中的化合物,它们可能会出现在最终药品中。大多数情况下,可浸出物属于提取物,但是由于与产品成分的相互作用,可能导致浸出物不属于可提取物。
▼ 对一次性系统生产组件提取研究的目的
目的是为了获得能在剧烈或极端条件下可提取物谱,并通过对可提取物的毒理学评估,帮助选择合适的组件系统,评估潜在的安全性风险。
可提取性研究还可作为基线,以考察SUS在一段时间内是否能保持一致性,例如对于重复使用的组件的评估,提取研究可为验证重复使用次数提供支持数据。
▼ 浸出研究的目的:
确定了在常规工艺条件下可能从 SUS向工艺溶液迁移的化合物,这些数据可以帮助毒理学家判断药物中是否含有可能危害患者安全的成分。
浸出物作为化学成份,可能与药品本身发生相互作用,浸出研究有助于评估药物的有效性和稳定性。
来源于SUS的浸出物会随时间持续存在,可能在稳定性研究中出现,对患者的安全和药物功效构成危险。
提取研究设计
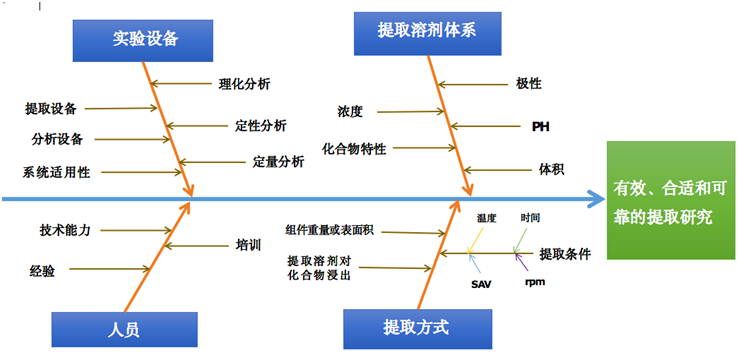
一次性组件提取试验设计应考虑影响提取质量和结果分析的因素:
▼ 提取溶剂系统
▼ 比表面积比
▼ 提取溶剂对关注可提取物的浸出力
▼ 提取温度和时间
▼ 一次性使用组件材质、结构
与材料相关的测试 基于易挥发物,半挥发物,非挥发物等目标物质特征,根据灵敏度和检测限选择合适的分析技术对提取物进行评价。
 ▼ 提取试验结果评估
▼ 提取试验结果评估
对可提取物进行毒理分析,识别并评估其潜在毒性及安全阈值。如果存在潜在安全风险,终端用户应主动与供应商沟通,暂时停止使用 SUS组件,直至实施风险缓解策略。
SUS供应商应根据行业公认标准采用通用提取方法,并以行业标准形式报告可提取物。终端用户应根据具体应用条件选择合适的提取条件和分析方法,以便对潜在浸出物进行准确预测。
浸出研究设计
SUS浸出物的评估应建立在风险管理基础之上,采用科学的方法,以确保最终药物产品的安全和质量。风险可能基于 SUS浸出物的安全风险程度、浸出的可能性和浸出物被检测的可能性。
同时,还需要考虑浸出物和药品化合物之间的相互作用,是否会影响药物的主要质量属性,如纯度、功效、特性,以及是否影响生产(例如细胞发酵过程)。
合作
SUS制造商、供应商和终端用户之间的合作和明确的角色定义对于确保患者安全和产品的有效性至关重要。
确定责任的最简单方法是建立清晰的沟通和透明的文件交换(认证、报告、结论、决策会议记录、任何法律协议)。利益相关者的责任可使用诸如 RACI (责任,责任,咨询,知情)责任分配表等工具确定。
SUS生产商在满足最终用户的期望(用户需求规范)方面投入了大量精力,应建立良好的沟通机制,使得最终用户和生产商共同参与到 SUS的风险评估中。
总之,提取研究的标准化和一致性,使得来自不同生产组件供应商的数据具有可比性,有利于组件之间的评估和比较。
对于浸出研究,我们则建议采用个案分析的方法,根据可提取数据,在毒理学家的监督下对特定产品进行评估,依照生产整体风险级别确定浸出研究开展程度。根据浸出的倾向性,通过风险评估和QbD原则,确定在哪个环节需要进行浸出研究,以避免不必要的浪费。
伯朗氏实验室(Brunslab Analytic)是一家国内领先的pre-clinical CRO公司,专门从事药包材相容性研究、医疗器械化学表征、包装材料及工艺组件可提取物和浸出物(E&L)研究,提供符合药典要求的分析服务和验证服务。
Brunslab Analytic以“超越客户的期望”并“确保顺利的合作”而自豪。我们的团队提供最全面的E&L研究,精通监管要求,可以在合规问题上为客户提供专业的支持。从委托项目的启动到完成,我们反应迅速、专业精神和追求细节。
随时联系我们,免费获取
-
最新的法规资料
-
咨询方案和报价
-
寻求其他合作
联系方式:
电话:+86 20 31068557
邮件:contact@brunslab.com