As the global Single Use System (SUS) market is about to break through the $15 billion mark, the sentiment in the pharmaceutical industry is not entirely optimistic. In our daily interactions with multiple pharmaceutical enterprises at Brunslab, we have heard an increasingly frequent term—”material compatibility anxiety.” This anxiety is not unfounded; it is quietly spreading from pharmaceutical R&D laboratories to procurement decision-making layers, and even affecting investors’ assessment logic for biopharmaceutical capacity expansion projects.
What explains the emergence of such collective anxiety behind a rapidly growing market? How will it reshape the entire pharmaceutical supply chain landscape? This article attempts to answer these two questions.
Hidden Concerns Behind Market Prosperity
The penetration speed of Single Use Systems in the biopharmaceutical field is remarkable. According to the latest research data from Coherent Market Insights, the global single-use bioreactor and supporting system market is expanding at a double-digit annual compound growth rate, pushing the overall SUS market size toward the $15 billion threshold.
**The logic behind this growth is clear**: single-use systems can significantly reduce cross-contamination risks between batches, shorten production line changeover times, and reduce cleaning validation costs—advantages that are nearly irreplaceable for CDMO services with multiple product types and small batch sizes.
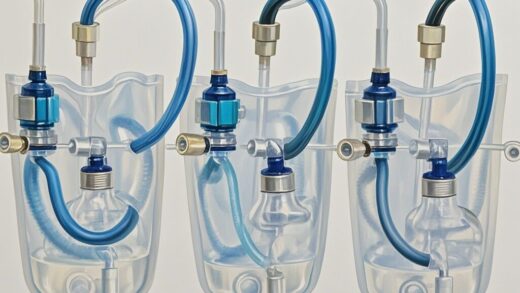
However, it is precisely this rapid expansion that has intensified the urgency of material compatibility issues. As SUS has progressively expanded from early cell culture stages to upstream fermentation, downstream purification, formulation filling, and even final packaging, pharmaceutical enterprises are no longer facing single-component material evaluation; instead, they must manage an entire supply chain composed of dozens of different material components.
Silicone tubing for single-use bioreactor bags, polyether sulfone membrane for filters, polypropylene connectors for fittings, multi-layer co-extruded films for storage bags—each material directly interacts with the drug product contact surface, and under long-term storage or specific process conditions, the extractables and leachables from these materials may become key variables affecting drug product safety.
A Often Overlooked Complexity: Structural Risks of Multi-Layer Supply Chains
To understand “material compatibility anxiety”, one must first comprehend the complexity of contemporary pharmaceutical supply chains. In traditional stainless steel systems, pharmaceutical enterprises have relatively concentrated control over contact materials—the compositional standards, polishing requirements, and surface passivation processes for 316L stainless steel are clearly specified, and suppliers are relatively stable.
However, in the SUS domain, a typical antibody drug production line may involve components supplied by more than 15 different suppliers, each with different raw material formulations, production processes, and quality control standards. Even products from different batches of the same supplier may exhibit extractables profile fluctuations due to subtle differences in raw material sources.
This complexity is evident in the participant list of the 2026 E&L Summit (Extractables & Leachables Summit). From the composition of sponsors and speakers, the institutions participating in E&L evaluation include globally leading third-party testing laboratories such as SGS, Eurofins, and Element Materials Technology, as well as pharmaceutical enterprises including Novartis, Sanofi, and Baxter Healthcare, along with component suppliers such as West Pharma and Becton Dickinson, and toxicological assessment and risk modeling professional institutions such as Lhasa Limited and Safetree Consulting. This vast ecosystem precisely illustrates that solving E&L problems requires multi-party collaboration, rather than any single entity working independently.
<p>In Brunslab’s actual projects, we have observed an increasing number of pharmaceutical enterprises requiring their SUS suppliers to provide complete material formulation information, not just Certificates of Conformity (CoC). Behind this trend lies the enterprises’ realization that compliance certificates can only prove that "the submitted sample passed a certain test standard," but cannot answer two fundamental questions: "How is batch-to-batch consistency guaranteed?" and "What will happen under specific drug formulation conditions?"
Regulatory Framework Evolution: From “Qualification Proof” to “Process Understanding”
If market expansion is the catalyst for “material compatibility anxiety,” then the evolution of regulatory frameworks provides a legitimate foundation for this anxiety. In recent years, a series of regulatory updates represented by ICH Q3E, USP <665>, and FDA guidance documents are fundamentally changing industry expectations for E&L evaluation.
The ICH Q3E guideline (Guidance for Industry: Evaluation and Control of Extractables/Leachables for Pharmaceutical Marketing Applications) is about to enter implementation phase. Its core concept shifts from “testing-based compliance” to “risk-based process understanding.” This means pharmaceutical enterprises can no longer simply rely on supplier-provided extractables data; they need to establish a deep understanding of the material-drug interaction mechanisms.
The revised version of USP <665> also emphasizes the individualized principle of risk assessment—when the same component is used for different administration routes, different contact durations, or different drug formulations, risk levels and required testing scopes may vary significantly.
This shift in regulatory philosophy presents a dual challenge to the industry. On one hand, it requires pharmaceutical enterprises to possess stronger materials science and chemical analysis capabilities, enabling them to design customized E&L research programs based on their product characteristics. On the other hand, it also requires the supply chain to have sufficient transparency, willing to share material formulation details with downstream customers and accept customer audits of their production processes.
Notably, regulatory agencies themselves are continuously adjusting their expectations. The participation of FDA representatives at the 2026 E&L Summit indicates that regulatory agencies will communicate directly with the industry on implementation details of the new guidelines. Referring to past experience, the initial period of any major regulatory framework adjustment is often accompanied by a wave of companies receiving deficiency letters due to understanding deviations or insufficient preparation. Those enterprises that have established systematic E&L management capabilities in advance will hold a significant competitive advantage.
Unknown Compound Issues: The Technical Deep Waters Faced by the Industry
Among all material compatibility-related technical challenges, the identification and risk assessment of unknown compounds is currently recognized as the industry’s greatest pain point.
Traditional GC-MS (Gas Chromatography-Mass Spectrometry) and LC-MS (Liquid Chromatography-Mass Spectrometry) analysis can identify most extractables, but there is typically still a certain percentage of compounds that cannot be matched by existing databases.
> These “unknowns” may be organic oligomers, catalyst residues, photoinitiator decomposition products, or new substances formed through inter-layer diffusion in multi-layer co-extruded films. Their chemical structures are unknown, toxicological data is missing, yet they may equally exist in the final drug product.
Facing this dilemma, some leading laboratories have begun exploring structural clustering methodologies. Nelson Labs’ three-phase framework is representative of this approach: Step one, define 12 categories of compounds of concern related to E&L; Step two, use high-resolution mass spectrometry and database comparison to preliminarily classify detected unknowns; Step three, conduct toxicological risk assessment based on category attribution. The advantage of this methodology is that even if the molecular structure of each unknown compound cannot be precisely defined, reasonable judgments about their safety risks can still be made.
However, implementing this methodology is no easy task. It requires laboratories to have strong cross-disciplinary capabilities in organic chemistry and toxicology, requires pharmaceutical enterprises to accept a certain degree of “approximate judgment” rather than “precise proof,” and also requires regulatory agencies to recognize the rationality of this risk assessment logic. In our project communications with clients, many enterprises are precisely stuck at this point—they can understand the value of this methodology, but within their internal decision-making chains, it is difficult to convince quality leaders to accept “risk assessment based on category inference” rather than “risk assessment based on precise identification results.”
Cost and Speed: A Trade-off That Cannot Be Avoided
The cost of material compatibility research cannot be ignored. A complete extractables and leachables study may cost between tens of thousands to hundreds of thousands of dollars, depending on product type and evaluation scope, and research cycles typically take 3 to 6 months. For biopharmaceutical projects in early clinical stages, this means additional capital investment and waiting time; for pharmaceutical enterprises with established mature production systems, it means every major process change may trigger a new round of E&L evaluation, thereby affecting capacity switchover flexibility.
Cost pressures have given rise to several coping strategies in the industry. Large pharmaceutical enterprises tend to establish internal E&L research capabilities or establish long-term cooperative frameworks with a few core testing laboratories to obtain more favorable pricing and faster scheduling. Smaller biotech companies more rely on “general-purpose” E&L data packages provided by suppliers, followed by supplementary evaluations based on their product characteristics. Some CDMO enterprises have begun to use “completed E&L data packages” as a differentiating competitive selling point, demonstrating the maturity of their supply chain management to clients.
However, there is a risk worth warning about: if the industry over-relies on standardized “general-purpose” data packages, it may overlook the most critical part of material compatibility issues—the leaching behavior under specific drug formulation conditions.
The same SUS component may exhibit completely different leaching kinetics in high-concentration salt solutions, acidic buffer systems, or organic solvent co-solvent systems. In multiple compatibility research projects at Brunslab, we have observed that the leachable lists for the same component under different formulation conditions can differ by more than 30%, indicating that a “one-size-fits-all” data package strategy has obvious limitations.
Reconstruction of Supply Chain Discourse Power
The spread of “material compatibility anxiety” is quietly changing the discourse power landscape of the pharmaceutical supply chain.
In the past, the relationship between component suppliers and pharmaceutical enterprises was relatively simple: suppliers provided products meeting specifications, and pharmaceutical enterprises purchased as needed. E&L evaluation was more regarded as an auxiliary process of quality release, rather than a core basis for supply chain selection. However, now an increasing number of pharmaceutical enterprises are incorporating E&L capabilities into their evaluation systems at the supplier screening stage—they want suppliers to not only provide compliance certificates but also complete material formulations, detailed extractables research data, capabilities for identifying and classifying unknowns, and support for leaching simulation studies specific to formulations.
This change places significantly higher demands on component suppliers. Some leading suppliers have already begun investing in building internal E&L research capabilities or establishing strategic partnerships with professional laboratories. However, for the large number of small and medium-sized suppliers, the threshold for such investment remains relatively high. The industry is undergoing a round of consolidation; those suppliers that can demonstrate their material management capabilities and provide transparent data packages will more easily gain favor from leading pharmaceutical enterprises.
From another perspective, this reconstruction of discourse power has also created opportunities for professional third-party service institutions. The importance of roles such as E&L testing laboratories, toxicological assessment agencies, and supply chain consulting companies is steadily increasing. The expansion of the 2026 E&L Summit’s scale and the diversification of participants are precisely a microcosm of this trend.
Looking Forward: Establishing Systematic Material Compatibility Management Capabilities
Based on the above analysis, the essence of “material compatibility anxiety” is the structural contradiction between the rapid expansion of the SUS market and the relatively lagging E&L management capabilities in the industry. This contradiction will not automatically resolve as market growth slows; instead, it may continue to intensify as regulatory standards tighten and new products and technologies are introduced.
For pharmaceutical enterprises, establishing systematic material compatibility management capabilities is no longer an optional “nice-to-have,” but a “necessary condition” concerning product launch and patient safety. This requires enterprises to incorporate E&L evaluation into consideration at the early stages of supply chain management, establish clear supplier qualification standards and data exchange agreements, invest in building internal technical team capabilities, or choose trustworthy external partners to share research workload.
For component suppliers, proactively embracing stricter material management requirements and providing more transparent data support will become the key to winning trust from top-tier clients. Those suppliers who still regard material formulations as “trade secrets” and refuse moderate disclosure may find themselves increasingly被动 in the face of increasingly strict supplier audits.
At Brunslab, we have observed a clear trend: more and more clients are beginning to regard E&L research as a standard环节 in the product development cycle that cannot be skipped, rather than a “temporary remedy” when encountering regulatory reviews. This cognitive shift indicates that the industry is moving from passive compliance toward active risk management.
The $15 billion SUS market still holds abundant opportunities. However, those enterprises that can first break through the bottleneck of material compatibility will occupy a more favorable position in future competitive landscapes.
Disclaimer: The content of this article is based on publicly available industry information and industry conference materials, provided for industry reference only and does not constitute any investment advice or business decision basis. Specific E&L evaluation programs should be professionally designed in conjunction with product characteristics, process conditions, and regulatory requirements.
FAQ
1. What are the primary challenges in material compatibility evaluation for Single Use Systems (SUS)?
The primary challenges include: (1) the complex multi-layer supply chain involving numerous suppliers with different material formulations; (2) the identification and risk assessment of unknown compounds that cannot be matched by existing databases; (3) the significant variability in extractables and leachables under different drug formulation conditions; (4) the high cost and long research cycles (3-6 months) for comprehensive E&L studies; and (5) the difficulty in ensuring batch-to-batch consistency of materials from different suppliers.
2. How is the regulatory landscape evolving for E&L evaluation in the pharmaceutical industry?
Regulatory frameworks are shifting from “testing-based compliance” to “risk-based process understanding.” Key developments include: ICH Q3E implementation focusing on understanding material-drug interaction mechanisms rather than relying solely on supplier data; USP <665> emphasizing individualized risk assessment based on administration route, contact duration, and drug formulation; and increased FDA engagement through industry forums to clarify implementation expectations. This evolution requires pharmaceutical enterprises to establish stronger internal material science capabilities and demand greater transparency from their supply chains.
3. What strategies can pharmaceutical companies adopt to manage material compatibility risks effectively?
Effective strategies include: (1) incorporating E&L evaluation considerations at the earliest stages of supply chain management; (2) establishing clear supplier qualification standards requiring complete material formulation information; (3) investing in internal technical capabilities or partnering with trusted external laboratories; (4) moving beyond “general-purpose” data packages to conduct formulation-specific leaching studies; (5) building long-term relationships with core testing laboratories for preferential pricing and faster turnaround; and (6) treating E&L research as a standard step in product development rather than a reactive measure during regulatory reviews.
Brunslab is a Chinese laboratory headquartered in Guangzhou, specializing in Extractables & Leachables (E&L) studies for pharmaceutical packaging and medical device materials. | Contact: Tel: +86 20 31068557 | Email: contact@brunslab.com